Medical Device Go-to-Market Strategy: The 2026 Operational Playbook
A medtech startup we worked with spent 18 months building a GTM plan that was really just a marketing plan with a regulatory timeline bolted on. They missed reimbursement coding entirely, got stonewalled by a hospital Value Analysis Committee on their second sales call, and burned through most of their seed funding before generating a single PO. That's the pattern we see over and over again: teams confuse a medical device go-to-market strategy with a marketing plan, when it's actually a regulatory + reimbursement + procurement strategy with marketing attached.
What You Need (Quick Version)
Five workstreams run in parallel, not in sequence: regulatory, clinical evidence, reimbursement, commercial, and operational. Most teams treat these as sequential steps and wonder why launch takes twice as long as planned.
Here's the timeline reality check: 64% of pre-commercial companies developing Class II/III devices expect the process to take 6+ years. But only 29% of companies that actually launched needed that long. The gap isn't about the FDA being slow. It's about teams planning poorly, running workstreams one after another, and discovering reimbursement requirements after they've already burned through their regulatory budget.
Five Parallel Workstreams
The biggest mistake in medtech launch planning is sequencing workstreams that should run simultaneously. Your regulatory submission, clinical evidence generation, reimbursement strategy, commercial model, and operational infrastructure aren't steps on a checklist. They're parallel tracks that inform each other continuously.
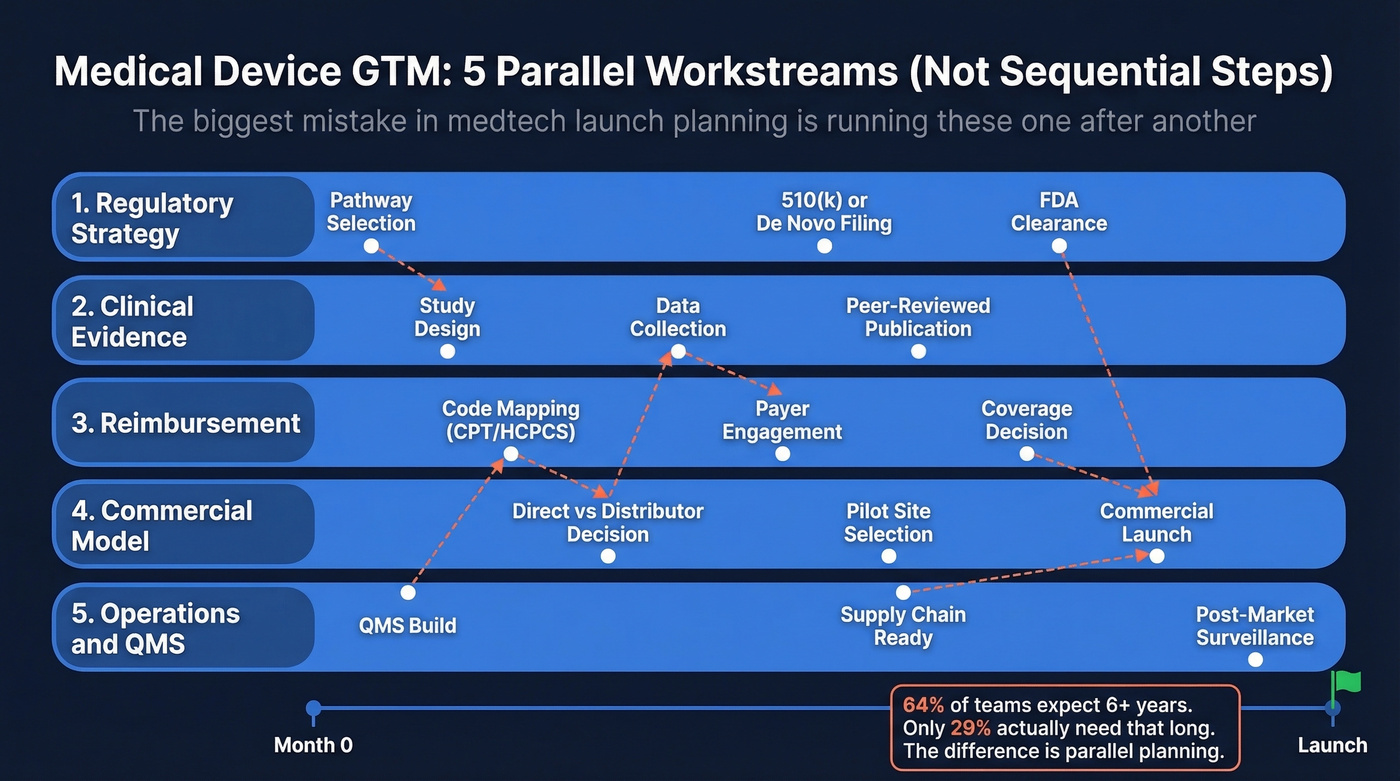
Regulatory decisions shape your clinical evidence requirements. Reimbursement coding determines how hospitals evaluate your financial ROI. Your commercial model dictates your Year 1 budget. And your operational infrastructure - QMS, supply chain, post-market surveillance - has to be ready before the first unit ships.
The companies that hit their launch windows plan all five from day one. The ones that slip by 12-24 months typically discover reimbursement or procurement requirements after regulatory clearance, then scramble to build evidence packets they should've started assembling two years earlier.
Choosing Your FDA Pathway
Your pathway choice is the single highest-leverage decision in your GTM timeline. It determines your user fees, evidence requirements, review duration, and how hospitals and payers perceive your device.
510(k) vs. De Novo vs. PMA
Roughly 70% of devices enter the US market via 510(k), which requires demonstrating substantial equivalence to a predicate device. De Novo is for novel, low-to-moderate risk devices with no predicate - a successful De Novo creates a new classification that future competitors can cite. PMA is the most rigorous path, reserved for high-risk Class III devices, requiring clinical trial data, design validation, and manufacturing inspections.
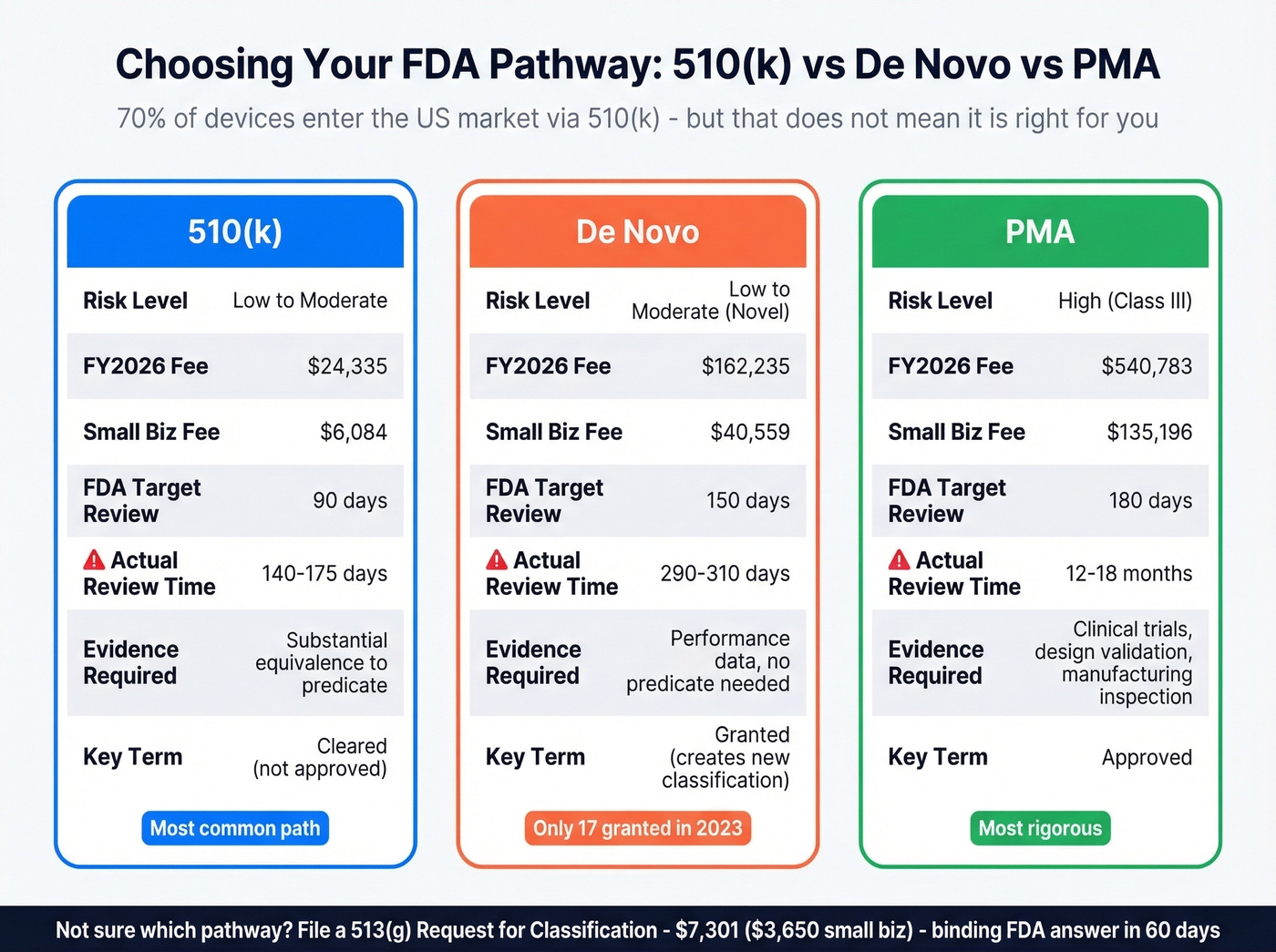
One critical distinction: 510(k) devices are "cleared," not "approved." PMA devices are "approved." Using "FDA approved" for a 510(k)-cleared device is illegal. Get this wrong in your marketing materials and you're looking at a warning letter.
Not sure which pathway applies? A 513(g) Request for Classification - $7,301, or $3,650 for small businesses - gets you a binding FDA determination within 60 days.
| 510(k) | De Novo | PMA | |
|---|---|---|---|
| FY2026 Fee | $24,335 | $162,235 | $540,783 |
| Small Biz Fee | $6,084 | $40,559 | $135,196 |
| Target Review | 90 days | 150 days | 180 days |
| Actual Review | 140-175 days | 290-310 days | 12-18 mo |
| Best For | Predicate exists | Novel, low-mod risk | High-risk Class III |
FDA user fees are updated annually. Budget for a 2-5% increase.
Why Your Timeline Is Wrong
Your board just asked when you'll have first revenue, and your 510(k) hasn't been filed yet. Recent CDRH data puts the average 510(k) clearance time at 140-175 days, with 70-80% of submissions exceeding the FDA's 90-day target. That's not a worst-case scenario. That's the norm.
CDRH has 2,260 staff processing 20,700+ submissions per year. In early 2025, 220+ jobs were eliminated. The growing share of AI-enabled submissions - 1,250+ authorized, up from fewer than 50 in 2010 - adds complexity to every reviewer's queue, stretching timelines further. For De Novo applications, expect 290-310 days. Only 17 De Novo requests were granted in 2023.
Regulatory Mistakes That Cost Years
Three patterns destroy timelines.
First, failing to develop a real regulatory strategy before starting submissions. Teams treat it as a form to fill out rather than a strategic framework. Second, treating the strategy as a static document. Markets shift, FDA guidance evolves, and your predicate landscape changes underneath you.
Third - and this one burns international expansion plans - assuming your US/EU strategy generalizes to other markets. We've watched companies lose 18+ months because they tried to copy-paste a 510(k) approach into an EU MDR submission. A one-size-fits-all approach to international submissions leads to multi-year delays and, in some cases, complete market access failure.
US-First or EU-First?
This used to be a real debate. It isn't anymore for most companies.
The US represents 46.4% of the global medtech market versus Europe's 26.4%. Since MDR/IVDR, choosing the EU as a first launch market has dropped roughly 40% among large IVD manufacturers and 33% among large device makers.
The bottleneck: 51 Notified Bodies across the EU have received 28,489 MDR applications but issued only 12,177 certificates - a throughput gap that isn't closing. For 60% of cases, the cycle from application to certificate runs 13-18 months, and 58% of that processing time is manufacturer-caused by incomplete submissions. MDR transition deadlines add urgency. Class III and implantable Class IIb devices must comply by December 31, 2027. Lower-risk devices have until December 31, 2028. IVD deadlines extend to December 31, 2029 on a risk-based schedule.
Unless you have a specific clinical or commercial reason to launch in Europe first - an existing distributor network, a clinical trial site advantage, or a faster reimbursement pathway in a specific country - default to US-first. The market is bigger, the regulatory pathway is more predictable even with CDRH staffing cuts, and you'll generate the revenue you need to fund EU expansion.
Reimbursement - The Step Everyone Skips
You can get FDA clearance, build a beautiful device, and still fail commercially because nobody figured out how hospitals get paid for using it. Reimbursement strategy runs parallel to regulatory - not after it.
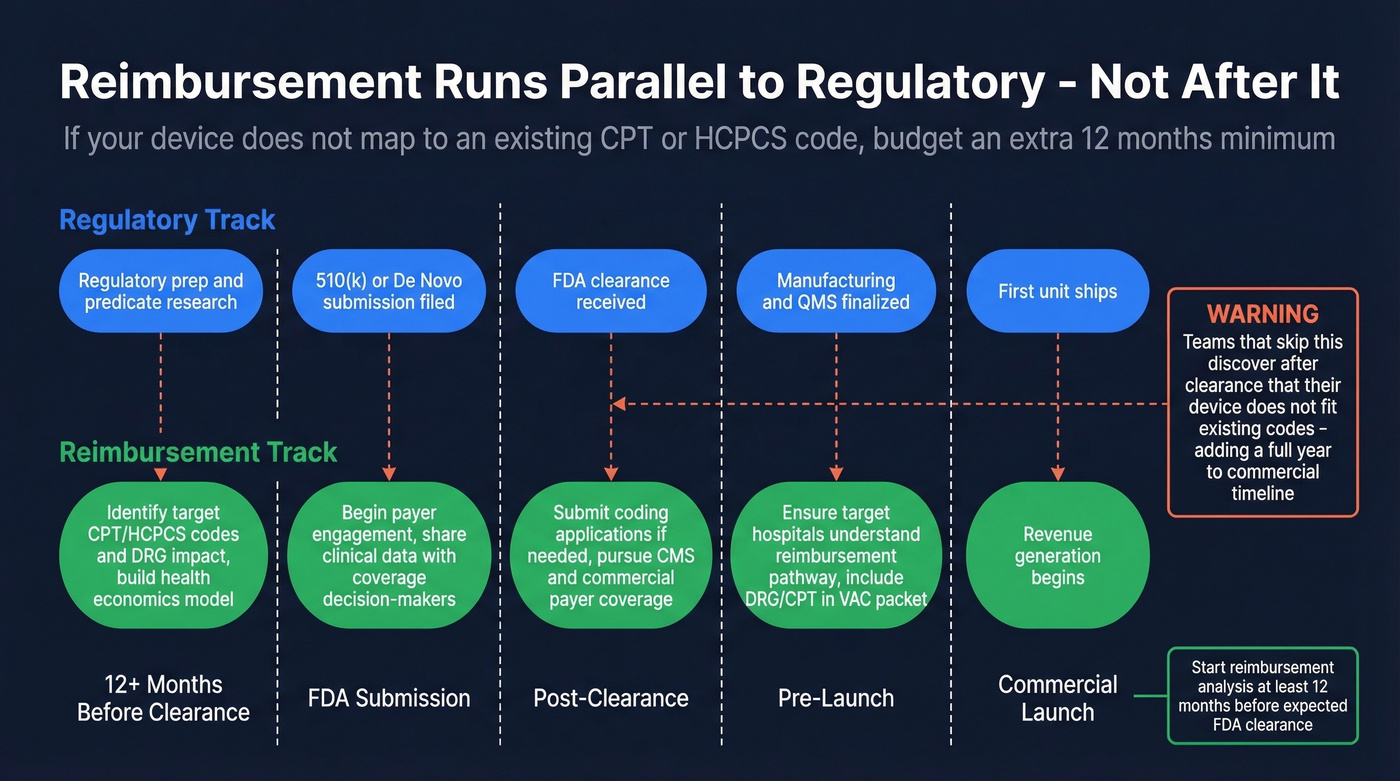
Here's the thing: if your device doesn't map to an existing CPT or HCPCS code, budget an extra 12 months minimum. No exceptions. A new code means coding applications, coverage determinations, and payer negotiations - and that clock doesn't start until you have clinical evidence to support the application.
The sequencing that works follows a clear progression. During regulatory prep, identify target CPT/HCPCS codes and DRG impact, then build your health economics model. At FDA submission, begin payer engagement and share clinical data with coverage decision-makers. Post-clearance, submit coding applications if needed and pursue coverage decisions from CMS and commercial payers simultaneously. Before commercial launch, ensure target hospitals understand the reimbursement pathway and include DRG/CPT details in your VAC evidence packet.
Teams that get this right start reimbursement analysis at least 12 months before expected FDA clearance. Teams that get it wrong discover - after clearance - that their device doesn't fit neatly into existing codes, and they've just added a year to their commercial timeline.
Your medtech GTM strategy is only as good as your ability to reach Value Analysis Committee members, procurement directors, and clinical champions. Prospeo's database has 300M+ profiles with 30+ filters - search by hospital system, department headcount, job title, and technographics to build targeted lists of the exact decision-makers who greenlight device purchases.
Stop getting stonewalled. Reach the right hospital buyers on the first attempt.
Hospital Procurement and the VAC
Your sales rep just got shut down by a hospital's Value Analysis Committee because you didn't have a cost-benefit analysis or a reimbursement code. This happens constantly, and it's entirely preventable.

VACs are formal hospital committees that evaluate every new product before a contract gets signed. They meet monthly or quarterly, which means missing one meeting cycle delays your approval by 30-90 days. Total approval timelines commonly run 3-6 months, and in larger health systems, up to a year.
The VAC evaluates clinical impact, cost justification, and operational fit. You need all three buttoned up before you walk in.
Clinical proof:
- Peer-reviewed studies or RCTs
- FDA clearance/approval documentation
- Comparative outcomes vs. current standard of care
Financial ROI:
- Cost-benefit analysis with total cost of ownership
- DRG/CPT reimbursement details showing the hospital won't lose money
- Volume projections tied to patient population data
Operational fit:
- Implementation and training plan with timeline
- EHR compatibility details, specifically HL7 and FHIR integration
- Support, warranty, and rollout case studies from pilot sites
In our experience, the internal champion makes or breaks the VAC process. You need a physician or department head who'll advocate for your device when you're not in the room. VACs often don't allow vendors to present directly - your champion has to answer every question the committee raises. "What does this replace?" "Is this must-have or nice-to-have?" "What's the patient volume?" That means arming them with a complete packet that anticipates objections. Skip the VAC meeting without a physician champion and you've wasted everyone's time.
Sales and Distribution Model
When Each Model Works
Go direct when your device is specialized or capital-intensive, volumes are localized, and clinical training is required for adoption. Direct gives you control over the sales narrative, but it's expensive and slow to scale.
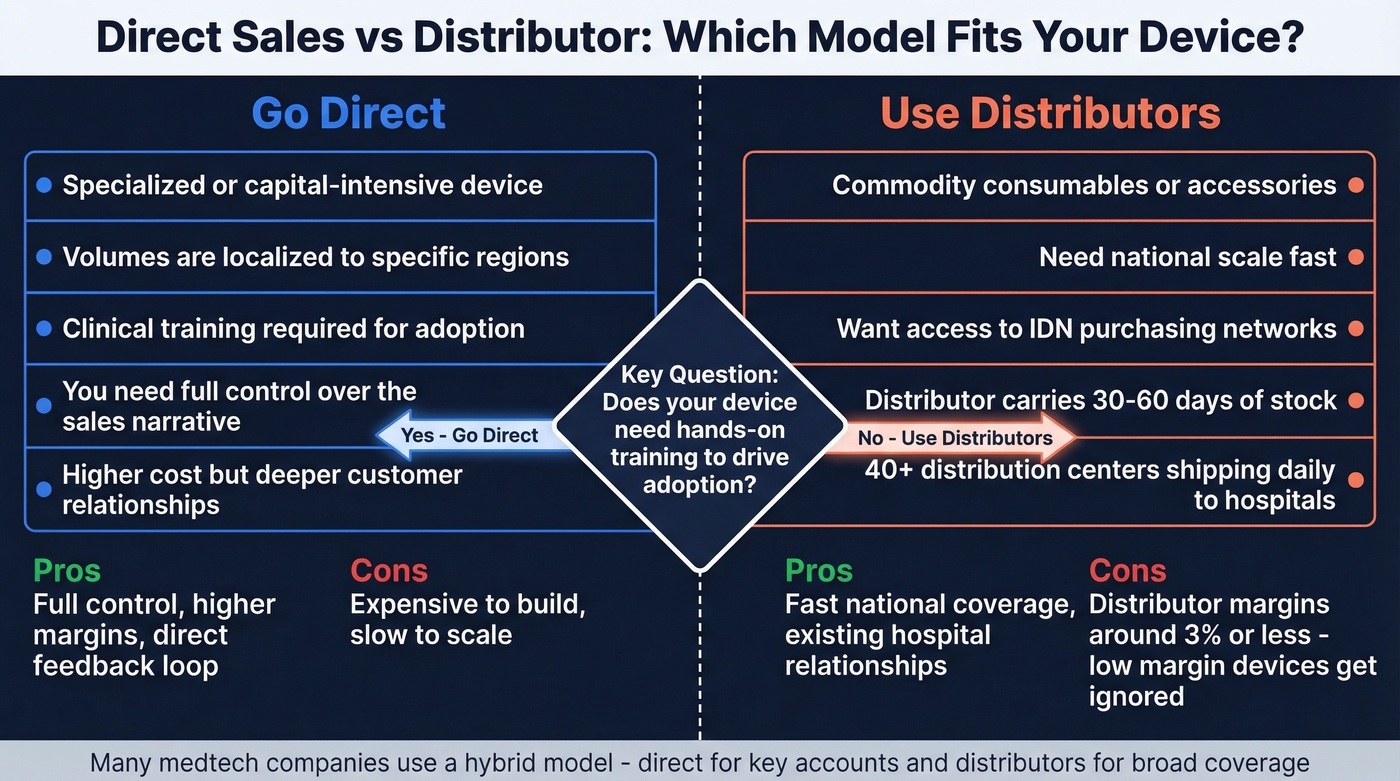
Use distributors when you're selling commodity consumables, need national scale fast, or want access to IDN purchasing networks. Distributors carry 30-60 days of stock and operate 40+ distribution centers shipping daily to hospitals. But their margins on distribution run roughly 3% or less - if your device doesn't generate meaningful margin for the distributor, it sits on the shelf.
Default to hybrid if you're a startup. We've seen early-stage companies burn through their first six months trying to go fully direct before realizing hybrid was the answer all along. Build direct relationships with your first 10-20 accounts to refine the sales process, then layer in distribution for geographic expansion.
One stat worth internalizing: 70%+ of HCPs now prefer virtual or hybrid touchpoints post-COVID. The field-sales-only model is increasingly misaligned with how clinicians want to engage.
Year 1 Budget Benchmarks
| Model | Year 1 Budget (EUR) | Team Size | Best For |
|---|---|---|---|
| Direct | EUR 300K-1M | 3-5 reps | Specialized devices |
| Distributor | EUR 100K-250K | 1-2 BD FTEs | Consumables at scale |
| Hybrid | EUR 150K-500K | Growth + med affairs | Most startups |
These are pre-Series A ranges. Post-Series B with aggressive growth targets, multiply accordingly.
Let's be honest: if your average deal size is under $25K, you probably don't need a direct sales force at all. A hybrid model with strong digital engagement and selective distribution will outperform five field reps burning cash on windshield time.
Marketing and Demand Generation
Channel ROI Benchmarks
Most medtech companies default to paid ads because results feel immediate. The data says that's the wrong instinct.
| Channel | Avg CAC | 3-Year ROI | Time to Results |
|---|---|---|---|
| SEO | $647 | 748% | 6-9 months |
| Speaking events | $518 | 856% | 2-4 weeks |
| $510 | 312% | 1-3 months | |
| PPC | $802 | 35% | Near instant |
Speaking engagements and SEO deliver 10-20x the 3-year ROI of PPC in medtech. If you're running a $50K/quarter marketing budget, put 60% into content and speaking, 25% into email nurture, and 15% into PPC for branded terms only.
KOL Strategy Beyond Conferences
Conference attendance is 35% below pre-pandemic levels. The old playbook - sponsor a booth, host a dinner, collect business cards - still works, but it reaches fewer people than it used to.
The digital KOL engagement model that's working now: record long-form interviews with your key opinion leaders for 30-60 minutes, then repurpose into short clips for professional networks and email nurture sequences. One 30-minute video testimonial can generate dozens of clips for retargeting campaigns that re-activate disengaged leads. Keep Sunshine Act compliance front of mind - document every interaction and payment.
For building your KOL outreach list, tools like Prospeo can pull verified contact information from conference speaker pages, hospital directories, or professional bios. At 98% email accuracy, your outreach actually lands instead of bouncing.
Building Your Target Account List
You've mapped your ICP: the 200 hospitals, the target specialties, the decision-maker titles. The strategy is solid. Now you need to actually reach these people, and this is where most medtech launch plans fall apart.
The gap between "have a sales strategy" and "execute outreach" is almost always a data problem. Your reps spend hours searching for contact information, cobble together emails from outdated conference lists, and send sequences that bounce at 15-25%. A B2B data platform with healthcare-specific filtering closes that gap - filter by job title, industry, company size, and department to build targeted lists of hospital decision-makers with verified emails refreshed weekly, not monthly.
Running five parallel GTM workstreams means your commercial team can't wait until clearance day to start building pipeline. With 98% verified email accuracy and 125M+ direct dials, Prospeo lets you map procurement contacts, KOLs, and clinical evaluators months before launch - so your first sales calls happen the week you get clearance, not months after.
Build your medtech pipeline before FDA clearance, not after.
Product Development Pitfalls
The engineering decisions you make in Year 1 can add 6-12 months to your GTM timeline. Material incompatibility alone accounts for 36% of medical device failures according to FDA data - selecting materials that can't withstand the sterilization method, use environment, or long-term performance requirements of the clinical setting.
Two other failure modes show up repeatedly. Poor EMC/EMI and electrical safety design creates problems that surface during verification testing and force redesign cycles. Mechanical designs that don't account for the clinical environment - cleaning chemicals, physical stress, daily handling by multiple users - fail in durability testing or, worse, in the field.
These are preventable. Early material testing under real use conditions, EMC testing before design freeze, and environmental stress testing that simulates actual clinical workflows catch most of these issues before they become 6-month delays. Cross-functional design reviews using ISO 14971's risk-based approach are the cheapest insurance you'll ever buy.
2026 Industry Benchmarks
The Greenlight Guru State of Medical Device report surveyed 500+ industry professionals, and the numbers are sobering:
- 56% still use paper-based or general-purpose tools for clinical data management
- 69% lack confidence their current QMS can support future growth
- 25% feel highly prepared for EU MDR compliance
- 16% feel prepared for QMSR requirements
- 33% are delaying new product development due to economic uncertainty
- 37% have implemented hiring freezes
The global medical device market was valued at over $560 billion in 2023 and is projected to reach $965 billion by 2032 - but the companies capturing that growth are the ones with operational maturity, not just innovative products. If your QMS is a collection of spreadsheets and your clinical data lives in paper binders, you're competing with one hand tied behind your back.
FAQ
How long does it take to bring a medical device to market?
Most Class II devices take 2-4 years from concept to commercial launch. The perception gap is real: 64% of pre-commercial companies expect 6+ years, but only 29% that launched actually needed that long. Parallel workstream planning is the biggest factor in compressing timelines.
What's the difference between FDA clearance and approval?
510(k) devices are "cleared" based on substantial equivalence to a predicate. PMA devices are "approved" based on clinical trial evidence. Using "FDA approved" for a 510(k)-cleared device is illegal in marketing materials and can trigger a warning letter.
How much does a 510(k) submission cost?
The FY2026 user fee is $24,335 standard or $6,084 for small businesses. Total submission costs including testing, consultants, and clinical data typically run $50K-$250K+ depending on device complexity and predicate availability.
Should I launch in the US or EU first?
Default to US-first in 2026. The US represents 46.4% of the global medtech market, and the EU MDR Notified Body bottleneck - 28,489 applications but only 12,177 certificates issued - makes European timelines unpredictable. Use early US revenue to fund EU expansion.